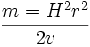تيتراسيون يعني شمارش
|
تيتراسيون يعني شمارش
همانند روش گراويمتري تيتراسيون نيز يكي از روشهاي قديمي در شيمي تجزيه مي باشد . كه هر دوي اين روشها مبتني بر واكنش شيميايي مي باشند.
در اين روش حجم محلول استاندارد ( تيترانت ) مصرفي، جهت واكنش كامل با ماده مجهول (آناليت) اندازه گيري مي شود كه اين حجم مبين مقدار ماده مجهول مي باشد. از آن زمان كه لوشميت و آووگادرو فهميدند كه يك گرم مولكول از ماده حاوي مقدار مشخصي ذره است محلول استاندارد ( تيترانت ) از حل كردن وزن مشخصي از ماده بدست آمد . اين بدان معني است كه اندازه گيري حجم مشخصي از تيترانت در طي تيتراسيون مي تواند شمارش تعداد مشخصي ذره از ماده مجهول ( آناليت ) باشد. بدين ترتيب تيتراسيون همان شمارش است . با وجود روشهاي دستگاهي و غير دستگاهي فيزيكي و شيميايي جديد امروزه هنوز تيتراسيون به عنوان يكي از روشهاي تجزيه اي كمي، در استانداردها جاي دارد كه اين حفظ موقعيت را مي توان به دلايل زير دانست . · سادگي انجام تيتراسيون : دستگاهها و روشها ساده بوده و به راحتي مهيا مي گردند . اساس تيتراسيون شناخته شده و به راحتي قابل يادگيري است. · تيتراسيون يكي از روشهاي قاطع براي اندازه گيري آناليت است و نتايج آزمايشات به صورت مستقيم مي تواند مقدار كمي ماده مجهول مورد اندازه گيري را تعيين كند اين روش به مانند روش هاي اسپكتروسكوپي يا فتومتري نياز به كاليبراسيون ندارد. · تيتراسيون به سرعت انجام مي پذيرد : زماني كه نتيجه گيري سريع براي شما مهم باشد اين روش مي تواند در وقت شما نسبت به روشهاي ديگر آناليز كمي، صرفه جويي قابل ملاحظه اي را انجام دهد. · تيتراسيون متضمن نتايج با صحت و دقت بالا مي باشد. معمولا" تكرار پذيري اكثر تيتراسيون ها كه با واحد انحراف استاندارد نسبي اندازه گيري مي شوند زير يك درصد مي باشند. · تيتراسيون قابليت اتوماسيون نيز دارد: تيتراسيون در حد بالايي قابليت اتوماسيون دارد اين بدان معناست كه در آناليز نمونه هاي روتين بسيار مناسب است · تيتراسيون خيلي مقرون بالصرفه است : در قياس با ساير روشهاي آناليز كمي، اين روش هزينه كم و كارايي بالايي دارد .
دسته بندي واكنشهاي تيتراسيون الف ) تيتراسيون هاي اسيد باز شامل : 1- اسيد و باز قوي 2- اسيد ضعيف با باز قوي 3- باز ضعيف با اسيد قوي 4- اسيد و باز ضعيف
ب ) تيتراسيون هاي رسوبي شامل : 1- تيتراسيون با تيترانت هاي غير آلي 2- تيتراسيون هاي سورفكتنتها
ج ) تيتراسيونهاي اكسيداسيون و احيا ( ردكس ) د ) تيتراسيون هاي كمپلكسومتري / كيليتومتري
روشهاي تشخيص نقطه اكي والان
اصولا" نقطه اكي والان زماني بدست مي آيد كه آناليت با تيترانت با نسب استكيومتري مربوطه به طور كامل واكنش دهند. اگر تيتراسيون را به عنوان " شمارش يونها يا مولكولها " بدانيم، بدست آوردن نقطه اكي والان بايد خيلي مهم باشد. انجام اين كار مي تواند با استفاده از خواص بعضي محلولها به عنوان شناساگر رنگي كه در بهترين حالت در نقطه اكي والان تغيير رنگ مي دهند، انجام شود. اما اين تغيير رنگ در نقطه پايان اتفاق مي افتد نه در نقطه اكي والان با اينكه نقطه پايان بسيار نزديك به نقطه اكي والان است . اما همواره در اين روش انتشار خطا خواهيم داشت از آن گذشته تشخيص افراد مختلف در تشخيص رنگ بسيار متفاوت است. اصولا" تشخيص نقطه اكي والان به سه روش مرسوم انجام مي پذيرد : - روش چشمي يا فتومتري - روش پتانسيومتري - روش بي ولتامتري/بي آمپرومتري در اين مبحث به روش پتانسيومتري كه روش مرسوم و متداول در اغلب استانداردها مي باشد مي پردازيم .
اساس روش پتانسيوتري :
روش پتانسيومتري يكي از متداول ترين روشهاي تشخيص نقطه اكي والان است اين روش تشخيص ، كل تيتراسيونهاي آبي ، غير آبي اسيد و باز ، اكسيداسيون احياء ، رسوبي و كمپلكسومتري را پوشش مي دهد . اساس تشخيص نقطه اكي والان به اين روش مبتني بر دو الكترود مي باشد ، يكي الكترود شناساگر و ديگري الكترود مرجع و چيزي كه در اينجا اندازه گيري مي شود پتانسيل الكترود ها نيست بلكه تفاضل پتانسيل بين الكترود شناساگر و الكترود كار در هر لحظه مي باشد. در واقع الكترود شناساگر پتانسيلي را تامين مي نمايد كه وابسته به وضعيت محلول داخل ظرف تيتراسيون مي باشد. و الكترود مرجع عهده دار تامين پتانسيلي است كه كاملا" مستقل از وضعيت محلول داخل ظرف تيتراسيون است . اين اختلاف پتانسيل بين دو الكترود توسط يك ولتامتر اندازه گيري مي شود كه مقاومت داخلي بسيار بالايي دارد.
رابطه تئوري اندازه گيري اين اختلاف پتانسيل به قرار زير مي باشد.
U = Uo + 2.303×RT ×Logai Zi × F
R= ثابت گازها ( I8.314 J/K/mol ) اختلاف پتانسيل بين دو الكترود =U پتانسيل الكترود شناساگر =Uo
دستگاههاي تيتراسيون هاي پتانسيومتري
همانطور كه در قبل نيز شرح داده شد اين روش نياز به مدار الكتريكي به نام ولتمتر دارد كه داراي مقاومت داخلي بالايي باشد.
امروزه دستگاههاي ولت متر ساخته شده براي اين منظور تنها يك ولتمتر ساده نيست . اين دستگاهها علاوه بر وظيفه اصلي كه به عهده دارند كه همانا پيدا كردن حجم مصرفي از تيترانت در نقطه اكي والان است بسيار پيشرفته تر شده ، مي توانند پس از پيدا كردن اين نقطه اطلاعات را نيز توسط فرمولي كه خود كاربران براي آنها تعريف مي كنند پردازش نمايند و نتيجه را به صورت مستقيم با ديمانسيون و تعداد ارقام پس از اعشار تعيين شده به نمايش در آورند . علاوه بر آن اطلاعات حاصل از هر آزمايش را مي توان توسط كامپيوتر ذخيره يا توسط دستگاه چاپگر نمود كه از اين طريق به كاربران اجازه مي دهند تا بانك اطلاعاتي تيتراسيون را در آزمايشگاه خود داشته باشد تا هر زمان كه نياز بود بتوانند به نتيجه آزمايشي در روز خاصي رجوع كرده آن را ملاحظه يا چاپ نمايد. در نسل جديد اين دستگاه كاربر قادر خواهد بود حتي تيترانت خود را در پله هاي i0.05µLit يكي از كمپاني هايي كه بيش از 65 سال در ارتباط با توليد اين نوع دستگاهها در جهان سابقه كار دارد كمپاني Metrohm سوئيس است كه در 65 سال پيش در شهر كوچكي در كشور سوئيس بوجود آمد و تا به و تا به امروز كه تبديل به بزرگترين كمپاني در اين ارتباط شده است . دستگاههايي كه در اين كمپاني امروزه براي تيتراسيون توليد مي كند ، در دو نسل Titrino,Titrando توليد مي شود. جديدترين مدل اين دستگاهها داراي قابليت هاي منحصر به فردي مي باشد كه در جهان بي همتاست استفاده از تكنولوژي ميكرو الكترو مكانيك، استفاده از بورتهاي هوشمند، استفاده از الكترود هوشمند، تبديل پتانسيل الكترود از آنالوگ به ديجيتال امكان انجام دو تيتراسيون به صورت همزمان از توانمندي هاي منحصر به فرد اين دستگاهها مي باشد. يكي ديگر از تيتراسيونهاي نام آشنا در صنايع نفت و گاز و پتروشيمي تيتراسيون كارل فيشر مي باشد در اين تيتراسيون كه I2 به عنوان تيترانت در تيتراسيوني با استكيومتري يك به يك با آب بعنوان آناليت واكنش مي دهد . و شما با اندازه گيري مقدار مصرف I2به آب نمونه مجهول پي مي بريد. كمپاني ياد شده بزرگترين و دقيق ترين اين دستگاهها را توليد مي نمايد از ديگر تيتراسيونهاي نام آشنا در صنعت نفت و گاز و پتروشيمي مي توان اندازه گيريH2S، مركاپتان ، برم نامبر ، برم ايندكس ،TBN،TAN را نام برد. كه در اين ارتباط مي توان از روشهاي از پيش آماده خود كمپاني نيز بهره گرفت . در پايان شركت سرمد طب مفتخر است تا با ارائه خدمات پس از فروش از قبل ارائه بولتن هاي كاربردي اطلاعات روز تهيه جزوات و خدمات كاليبراسيون و تامين و قطعات دستگاههاي ياد شده هر چند، گام كوچكي در راستاي اعتلاي اين بخش از صنعت كشور بردارد |
|
تيتراسيون يعني شمارش
همانند روش گراويمتري تيتراسيون نيز يكي از روشهاي قديمي در شيمي تجزيه مي باشد . كه هر دوي اين روشها مبتني بر واكنش شيميايي مي باشند.
در اين روش حجم محلول استاندارد ( تيترانت ) مصرفي، جهت واكنش كامل با ماده مجهول (آناليت) اندازه گيري مي شود كه اين حجم مبين مقدار ماده مجهول مي باشد. از آن زمان كه لوشميت و آووگادرو فهميدند كه يك گرم مولكول از ماده حاوي مقدار مشخصي ذره است محلول استاندارد ( تيترانت ) از حل كردن وزن مشخصي از ماده بدست آمد . اين بدان معني است كه اندازه گيري حجم مشخصي از تيترانت در طي تيتراسيون مي تواند شمارش تعداد مشخصي ذره از ماده مجهول ( آناليت ) باشد. بدين ترتيب تيتراسيون همان شمارش است . با وجود روشهاي دستگاهي و غير دستگاهي فيزيكي و شيميايي جديد امروزه هنوز تيتراسيون به عنوان يكي از روشهاي تجزيه اي كمي، در استانداردها جاي دارد كه اين حفظ موقعيت را مي توان به دلايل زير دانست .
· سادگي انجام تيتراسيون : دستگاهها و روشها ساده بوده و به راحتي مهيا مي گردند . اساس تيتراسيون شناخته شده و به راحتي قابل يادگيري است. · تيتراسيون يكي از روشهاي قاطع براي اندازه گيري آناليت است و نتايج آزمايشات به صورت مستقيم مي تواند مقدار كمي ماده مجهول مورد اندازه گيري را تعيين كند اين روش به مانند روش هاي اسپكتروسكوپي يا فتومتري نياز به كاليبراسيون ندارد. · تيتراسيون به سرعت انجام مي پذيرد : زماني كه نتيجه گيري سريع براي شما مهم باشد اين روش مي تواند در وقت شما نسبت به روشهاي ديگر آناليز كمي، صرفه جويي قابل ملاحظه اي را انجام دهد. · تيتراسيون متضمن نتايج با صحت و دقت بالا مي باشد. معمولا" تكرار پذيري اكثر تيتراسيون ها كه با واحد انحراف استاندارد نسبي اندازه گيري مي شوند زير يك درصد مي باشند. · تيتراسيون قابليت اتوماسيون نيز دارد: تيتراسيون در حد بالايي قابليت اتوماسيون دارد اين بدان معناست كه در آناليز نمونه هاي روتين بسيار مناسب است · تيتراسيون خيلي مقرون بالصرفه است : در قياس با ساير روشهاي آناليز كمي، اين روش هزينه كم و كارايي بالايي دارد .
دسته بندي واكنشهاي تيتراسيون الف ) تيتراسيون هاي اسيد باز شامل : 1- اسيد و باز قوي 2- اسيد ضعيف با باز قوي 3- باز ضعيف با اسيد قوي 4- اسيد و باز ضعيف
ب ) تيتراسيون هاي رسوبي شامل : 1- تيتراسيون با تيترانت هاي غير آلي 2- تيتراسيون هاي سورفكتنتها
ج ) تيتراسيونهاي اكسيداسيون و احيا ( ردكس ) د ) تيتراسيون هاي كمپلكسومتري / كيليتومتري
روشهاي تشخيص نقطه اكي والان اصولا" نقطه اكي والان زماني بدست مي آيد كه آناليت با تيترانت با نسبت استكيومتري مربوطه به طور كامل واكنش دهند. اگر تيتراسيون را به عنوان " شمارش يونها يا مولكولها " بدانيم، بدست آوردن نقطه اكي والان بايد خيلي مهم باشد. انجام اين كار مي تواند با استفاده از خواص بعضي محلولها به عنوان شناساگر رنگي كه در بهترين حالت در نقطه اكي والان تغيير رنگ مي دهند، انجام شود. اما اين تغيير رنگ در نقطه پايان اتفاق مي افتد نه در نقطه اكي والان با اينكه نقطه پايان بسيار نزديك به نقطه اكي والان است . اما همواره در اين روش انتشار خطا خواهيم داشت از آن گذشته تشخيص افراد مختلف در تشخيص رنگ بسيار متفاوت است.
اصولا" تشخيص نقطه اكي والان به سه روش مرسوم انجام مي پذيرد : - روش چشمي يا فتومتري - روش پتانسيومتري - روش بي ولتامتري/بي آمپرومتري
در اين مبحث به روش پتانسيومتري كه روش مرسوم و متداول در اغلب استانداردها مي باشد مي پردازيم . اساس روش پتانسيوتري : روش پتانسيومتري يكي از متداول ترين روشهاي تشخيص نقطه اكي والان است اين روش تشخيص ، كل تيتراسيونهاي آبي ، غير آبي اسيد و باز ، اكسيداسيون احياء ، رسوبي و كمپلكسومتري را پوشش مي دهد . اساس تشخيص نقطه اكي والان به اين روش مبتني بر دو الكترود مي باشد ، يكي الكترود شناساگر و ديگري الكترود مرجع و چيزي كه در اينجا اندازه گيري مي شود پتانسيل الكترود ها نيست بلكه تفاضل پتانسيل بين الكترود شناساگر و الكترود كار در هر لحظه مي باشد. در واقع الكترود شناساگر پتانسيلي را تامين مي نمايد كه وابسته به وضعيت محلول داخل ظرف تيتراسيون مي باشد. و الكترود مرجع عهده دار تامين پتانسيلي است كه كاملا" مستقل از وضعيت محلول داخل ظرف تيتراسيون است . اين اختلاف پتانسيل بين دو الكترود توسط يك ولتامتر اندازه گيري مي شود كه مقاومت داخلي بسيار بالايي دارد.
رابطه تئوري اندازه گيري اين اختلاف پتانسيل به قرار زير مي باشد
U = Uo + 2.303×RT ×Logai Zi × F
R= ثابت گازها ( 8.314 J/K/mol ) U= اختلاف پتانسيل بين دو الكترود Uo= پتانسيل الكترود شناساگر Zi= بار يوني F= عدد فارادي ( 96484.56 C/mol ) ai= فعاليت يوني
دستگاههاي تيتراسيون هاي پتانسيومتري
همانطور كه در قبل نيز شرح داده شد اين روش نياز به مدار الكتريكي به نام ولتمتر دارد كه داراي مقاومت داخلي بالايي باشد.
امروزه دستگاههاي ولت متر ساخته شده براي اين منظور تنها يك ولتمتر ساده نيست . اين دستگاهها علاوه بر وظيفه اصلي كه به عهده دارند كه همانا پيدا كردن حجم مصرفي از تيترانت در نقطه اكي والان است بسيار پيشرفته تر شده ، مي توانند پس از پيدا كردن اين نقطه اطلاعات را نيز توسط فرمولي كه خود كاربران براي آنها تعريف مي كنند پردازش نمايند و نتيجه را به صورت مستقيم با ديمانسيون و تعداد ارقام پس از اعشار تعيين شده به نمايش در آورند . علاوه بر آن اطلاعات حاصل از هر آزمايش را مي توان توسط كامپيوتر ذخيره يا توسط دستگاه چاپگر نمود كه از اين طريق به كاربران اجازه مي دهند تا بانك اطلاعاتي تيتراسيون را در آزمايشگاه خود داشته باشد تا هر زمان كه نياز بود بتوانند به نتيجه آزمايشي در روز خاصي رجوع كرده آن را ملاحظه يا چاپ نمايد. در نسل جديد اين دستگاه كاربر قادر خواهد بود حتي تيترانت خود را در پله هاي i0.05µLit اضافه نمايد .يكي از كمپاني هايي كه بيش از 65سال در ارتباط با توليد اين نوع دستگاهها در جهان سابقه كار دارد كمپاني Metrohm سوئيس است كه در 65 سال پيش در شهر كوچكي در كشور سوئيس بوجود آمد و تا به امروز كه تبديل به بزرگترين كمپاني در اين ارتباط شده است . دستگاههايي كه در اين كمپاني امروزه براي تيتراسيون توليد مي كند ، در دو نسل Titrando, Titrino توليد مي شوند .
جديدترين مدل اين دستگاهها داراي قابليت هاي منحصر به فردي مي باشد كه در جهان بي همتاست استفاده از تكنولوژي ميكرو الكترو مكانيك، استفاده از بورتهاي هوشمند، استفاده از الكترود هوشمند، تبديل پتانسيل الكترود از آنالوگ به ديجيتال امكان انجام دو تيتراسيون به صورت همزمان از توانمندي هاي منحصر به فرد اين دستگاه ها مي باشد. يكي ديگر از تيتراسيونهاي نام آشنا در صنايع نفت و گاز و پتروشيمي تيتراسيون كارل فيشر مي باشد در اين تيتراسيون كه I2 به عنوان تيترانت در تيتراسيوني با استكيومتري يك به يك با آب بعنوان آناليت واكنش مي دهد و شما با اندازه گيري مقدار مصرف I2 به آب نمونه مجهول پي مي بريد. كمپاني ياد شده بزرگترين و دقيق ترين اين دستگاهها را توليد مي نمايد از ديگر تيتراسيونهاي نام آشنا در صنعت نفت و گاز و پتروشيمي مي توان اندازه گيري H2S، مركاپتان ، برم نامبر ، برم ايندكس TAN,TBN را نام برد. كه در اين ارتباط مي توان از روشهاي از پيش آماده خود كمپاني نيز بهره گرفت . در پايان شركت سرمد طب مفتخر است تا با ارائه خدمات پس از فروش از قبل ارائه بولتن هاي كاربردي اطلاعات روز تهيه جزوات و خدمات كاليبراسيون و تامين و قطعات دستگاههاي ياد شده هر چند، گام كوچكي در راستاي اعتلاي اين بخش از صنعت كشور بردارد.
|